Hey, brother, align this!
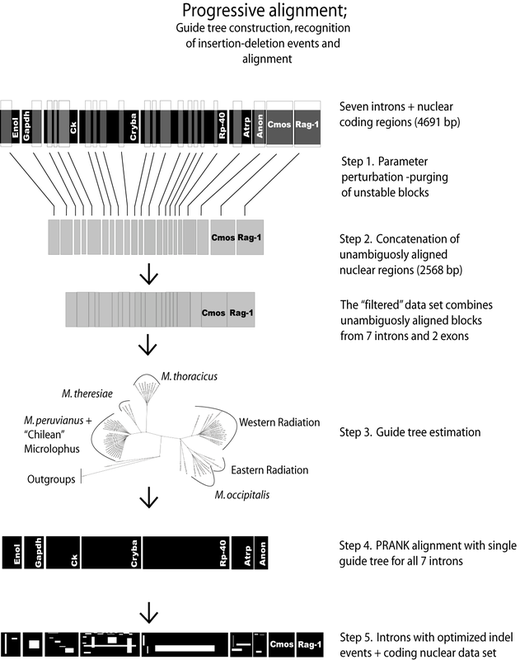
The alignment of homologous gene regions can be tricky. In particular if these genes correspond to non-coding regions. The figure to the right shows a set of homologous sequences from highly variable nuclear introns. To align this introns manually you need to know that not all gaps are created the same. Non-coding regions will naturally, accumulate both point and length mutations - gaps.
As stated above, not all gaps are the same. Indeed, insertions and deletions should be characterized as apples and oranges.
The two types of lenght mutations are vastly different. In fact, they do occur at very different frequencies, which is a strong hint of some type of selection acting against insertions across vertebrate genomes.
Notably, most alignment programs (e.g., CLUSTAL, MUSCLE, MAFFT, etc etc) will use a single penalization to place any indel in a given alignment. This (computationally feasible) alternative carries the assumption that the two mutational events are equivalent i.e., they are equally frequent.
I have suggested an empirical protocol, to enhance the application of an alignment program (PRANK; Loytinoja and Goldman, 2005PNAS; 2008Science [Excellent paper by the way]) that uses different gap penalties for insertions and deletions. Normally, both, alignment and phylogenetic analyses should be optimized at the same time. However, this is not an easy computational problem.
As new genomic data sets emerge, adequate alignment of indels will be key to phylogenetic reconstruction but also increasingly complex homology statements. Indels are prevalent and valuable source of information. Of course, one tends to discard data for which no clear homology statements can be made. But again, that does not mean a solution is not there. Perhaps, a few detours are needed.
As stated above, not all gaps are the same. Indeed, insertions and deletions should be characterized as apples and oranges.
The two types of lenght mutations are vastly different. In fact, they do occur at very different frequencies, which is a strong hint of some type of selection acting against insertions across vertebrate genomes.
Notably, most alignment programs (e.g., CLUSTAL, MUSCLE, MAFFT, etc etc) will use a single penalization to place any indel in a given alignment. This (computationally feasible) alternative carries the assumption that the two mutational events are equivalent i.e., they are equally frequent.
I have suggested an empirical protocol, to enhance the application of an alignment program (PRANK; Loytinoja and Goldman, 2005PNAS; 2008Science [Excellent paper by the way]) that uses different gap penalties for insertions and deletions. Normally, both, alignment and phylogenetic analyses should be optimized at the same time. However, this is not an easy computational problem.
As new genomic data sets emerge, adequate alignment of indels will be key to phylogenetic reconstruction but also increasingly complex homology statements. Indels are prevalent and valuable source of information. Of course, one tends to discard data for which no clear homology statements can be made. But again, that does not mean a solution is not there. Perhaps, a few detours are needed.
